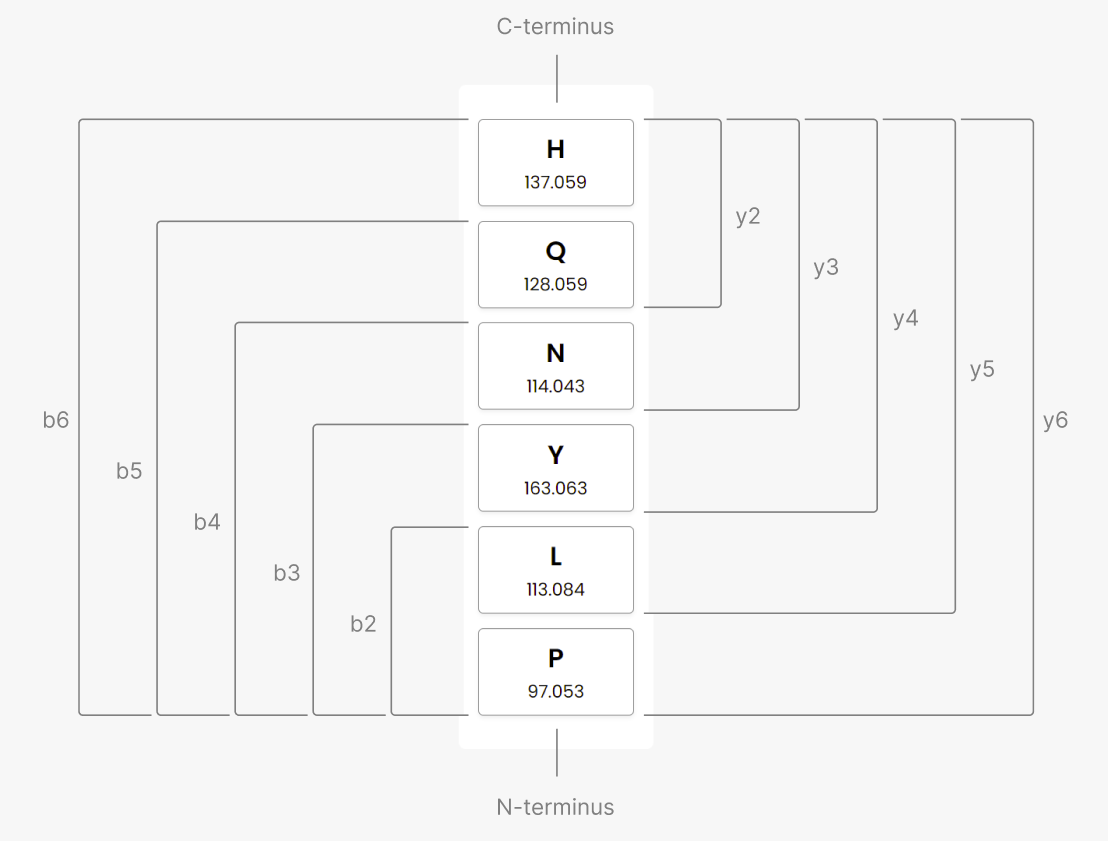
Peptide Fragments
Each bar represents the summed masses of a peptide fragment. The fragments range in length from dipeptides (length 2) to the entire peptide. All fragments include the C-terminus and/or N-terminus. The masses are arranged in ascending order, which does not necessarily alternate C- and N-terminus fragments: b2, y2, b3, y3, etc.
Close